ok: Hội chứng người hóa đá một căn bệnh di truyền hiếm gặp
Hội chứng SMS (Stone Man Syndrome) hay Người hóa đá hoặc gọi theo chuyên môn FOP (Fibrodisplasia ossificans progressiva) là căn bệnh di truyền hiếm gặp, xếp thứ nhất danh sách top 10 bệnh quái dị ít được nhắc đến, hiện trên thế giới có khoảng 800 người đã được xác nhận là mắc bệnh.
Vì sao FOP lại là bệnh quái dị?
Theo Từ điển y học trực tuyến thì FOP là căn bệnh mang tính di truyền cực hiếm, lần đầu tiên được bác sĩ người Pháp guy Patin phát hiện ra năm 1692, được ông mô tả là "cứng như gỗ". FOP thường xuất hiện ở trước tuổi dậy thì trong đó các mô liên kết bị xơ cứng triệt để và phát sinh tình trạng tạo xương các bộ phận như dây chằng, cơ mô liên kết, gân và da. Sự phát triển bất thường của xương có thể dẫn đến tình trạng cứng hóa, hạn chế việc chuyển động của các khớp như đầu gối, cổ tay, vai cột sống và cổ. Nếu nặng có thể làm tê liệt, biến cơ thể thành khối giống như một bức tượng, như nhân vật trong phim Clash of the Titans (Cuộc chiến giữa các vị thần). Nghĩa là FOP tạo ra thêm nhiều xương mới ngay trong các mô liên kết của cơ thể, vì vậy, FOP mới được dịch là "các mô liên kết mềm dần dần biến thành xương", một dạng bệnh cực lạ, trong đó một bộ phận thường là mô mềm chuyển hóa thành một bộ phận hoàn toàn khác, đó là xương. Theo số liệu thống kê, trung bình cứ 2 triệu người thì có 1 mắc bệnh, tức khoảng trên 3.300 người lâm bệnh nhưng nay mới chỉ có 700 - 800 ca được khám và chữa bệnh.
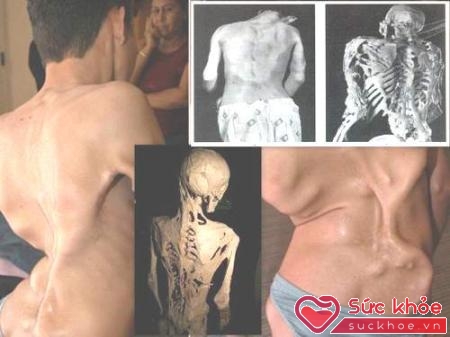
Bệnh có thể khiến người bệnh bị co cứng, bất động.
Hai trường hợp mắc bệnh FOP mới nhất vừa được dư luận nhắc đến là một bé gái 6 tuổi ở Bellevue, bang Ohio, Mỹ tên là Ali McKean. Căn bệnh này làm cho cơ thể Ali căng cứng, không di chuyển hay cử động được, giống như một bức tượng. "Ali đang dần cứng hóa và đóng băng hoàn toàn", chị Angela, mẹ của Ali cho biết. Trường hợp thứ hai là một thiếu nữ Seanie Nammock, 17 tuổi, ở vùng Tây Luân Đôn, Anh. Do mắc bệnh FOP di truyền nên cổ tay Seanie bị co cứng và bất động, xương hai bên bả vai bị tê liệt hoàn toàn. Bất kỳ va chạm nhỏ nào cũng có thể gây đau đớn, bởi vậy, mọi sinh hoạt của Seanie đều phải nhờ vào bố mẹ và những người xung quanh.
Triệu chứng ban đầu của bệnh FOP
Thông thường, ở nhóm trẻ mắc bệnh FOP thì có tới 50% tự kế thừa, đây là căn bệnh bẩm sinh phát triển ngay từ khi còn nằm trong bụng mẹ. Dấu hiệu dễ nhận biết như biến dạng ngón chân, đặc biệt ngón chân cái bị ngắn, co quắp vào trong (khoảng 50% số ca mắc bệnh), cột sống bị cứng. Ngoài ra, những đứa trẻ mắc bệnh này thường có các dị tật ở khuỷu khớp nối các xương sườn với cột sống, hạn chế phát triển vòng ngực làm cho việc hô hấp gặp nhiều khó khăn. Nếu xuất hiện sớm, trẻ không thể bò được mà thường phải kéo lê người.
Theo năm tháng, bệnh FOP phát triển, xuất hiện các khối u mềm hoặc các khối u ở cổ, lưng và vai, từ đây, nó tạo ra những xương mới tại các bộ phận này. Một bộ phận xương mới phải mất khoảng 8 tuần để phát triển, người bệnh xuất hiện tình trạng sốt nhẹ và đau viêm nhiễm, sưng tấy khó chịu. Khi lên 10 tuổi, những đứa trẻ mắc bệnh thường nhận thức được căn bệnh và cố gắng để tồn tại. Do chứa đựng nhiều bí ẩn nên khoa học vẫn chưa hiểu hết nguyên nhân gây bệnh. Người ta mới chỉ tình nghi đến những nguy cơ như do tiêm bắp, do các thủ thuật về răng lợi, do virut cúm, do té ngã... Căn bệnh phát triển thêm xương mới này thường diễn ra từ đầu đến chân, từ phía trước cho đến phía sau, nghĩa là ảnh hưởng đến toàn bộ cơ thể. Bệnh ngày càng phát triển nên đến khoảng 20 - 30 tuổi, người bệnh mất dần khả năng đi lại, thậm chí còn gặp khó khăn khi cử động nên thường phải nằm liệt và trông chờ vào sự giúp đỡ của người thân. Ngoài ra, có tới 50% số người mắc bệnh FOP bị điếc, bất động và ảnh hưởng đến chức năng hô hấp 95% bệnh nhân FOP bị ảnh hưởng chức năng tim mạch.
Năm 2006, nhóm chuyên gia ở ĐH Pennsylvania Mỹ do tiến sĩ F. Kaplan đứng đầu đã nghiên cứu và tìm ra một đột biến trong thụ thể A của gen ACVR1 týp 1 hay còn gọi là ALK2 (activin receptor-like kinase 2), thành viên thuộc nhóm protein có tên BMP týp 1 (Bone morphogenetic protein type I receptors). Theo đó, những ai có cả hai copy gen ACVR1/ALK2 trong các tế bào cơ thể thì rủi ro mắc bệnh FOP càng lớn.
Chẩn đoán, chữa trị
Theo Hiệp hội FOP quốc tế, có tới 87% số ca mắc bệnh là bị chẩn đoán nhầm, chỉ có 8% là chẩn đoán chính xác, 10% bác sĩ mới được nghe qua về căn bệnh này, 68% điều trị không thích hợp, thậm chí có tới 49% bị thiểu năng do điều trị không đúng cách gây ra.
- Dịch bệnh không chừa một ai, điểm khác giữa mắc Covid-19 và... (Chủ nhật, 20:19:06 16/05/2021)
- 6 cơn đau thường gặp báo hiệu cơ thể đang gặp nguy hiểm, bị... (Thứ bảy, 12:57:06 08/05/2021)
- 7 dấu hiệu ở bàn chân cảnh báo cơ thể đang gặp nguy, cần... (Thứ sáu, 16:40:02 07/05/2021)
- 5 dấu hiệu cảnh báo sớm ung thư tuyến tụy chớ dại bỏ qua (Thứ Ba, 15:28:00 04/05/2021)
- Cơ thể có 5 dấu hiệu này chứng tỏ bạn đang thiếu vitamin C,... (Thứ bảy, 10:43:03 01/05/2021)
- Chân sưng phù: Đừng xem thường bởi đó là dấu hiệu của 7... (Thứ bảy, 21:06:08 24/04/2021)
- 2 món không ăn buổi sáng, 2 thứ không đụng buổi tối, 3 điều... (Thứ Ba, 08:40:06 20/04/2021)
- Buổi sáng ngủ dậy thấy 4 việc này chứng tỏ bệnh gan bạn... (Chủ nhật, 08:30:03 18/04/2021)
- Tê tay tưởng chuyện thường nhưng cẩn thận, đó là dấu hiệu... (Thứ năm, 13:21:02 15/04/2021)
- Đừng coi thường dấu hiệu cứng ngón tay, rất có thể đó là... (Thứ bảy, 08:32:01 10/04/2021)
-
 Bí quyết sống khỏe
Bí quyết sống khỏe
Thứ năm, 14:36:38 09/07/2020
-
 Món ngon giữ lửa yêu
Món ngon giữ lửa yêu
Thứ năm, 14:35:20 09/07/2020
-
 Bệnh trẻ em
Bệnh trẻ em
Thứ năm, 14:33:40 09/07/2020
-
 Bệnh phụ nữ
Bệnh phụ nữ
Thứ năm, 14:31:58 09/07/2020
-
 Bài thuốc hay
Bài thuốc hay
Thứ năm, 14:30:50 09/07/2020
-
 Đồ Chơi Pop it bóp bóp Bấm Nút Bóp Bóng - Pop It
Đồ Chơi Pop it bóp bóp Bấm Nút Bóp Bóng - Pop It
Thứ tư, 20:15:08 15/02/2023
-
 Đồ chơi bộ hộp sét 6 món ô tô máy bay chạy cót xịn xò
Đồ chơi bộ hộp sét 6 món ô tô máy bay chạy cót xịn xò
Thứ Hai, 18:18:07 13/02/2023
-
 Máy gắp thú bông cho bé cỡ lớn BBT GLOBAL
Máy gắp thú bông cho bé cỡ lớn BBT GLOBAL
Chủ nhật, 15:54:03 12/02/2023